Retinoblastoma
Everything you need to know about retinoblastoma
Retinoblastoma is a primary malignant tumor of the retina. It is the most common intraocular cancer in children.
The occurrence of retinoblastoma is linked to a mutation in the RB1 gene, a tumor suppressor gene. This mutation can arise randomly in a precursor retinal cell but may also be present constitutionally (from birth) in all or part of the child’s cells. Retinoblastoma occurs in about half of cases in the context of a genetic predisposition.
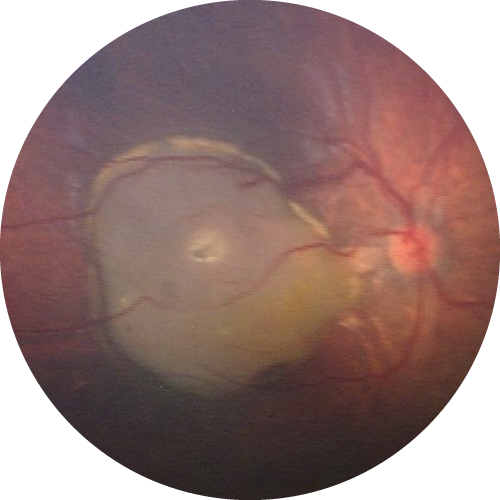
Except for children monitored from birth as part of a personalized surveillance program, the diagnosis is often delayed, typically identified by the appearance of a white pupil (leukocoria) or strabismus, which requires an urgent ophthalmologic examination with a dilated fundus exam. The diagnosis is then confirmed at a reference center at the Institut Curie in Paris. In cases of genetic predisposition, both eyes may be affected.
Early diagnosis determines the implementation of treatments that preserve the eyes and maintain the best possible vision according to the initial involvement. In some cases, treatment may require removal of the eye (enucleation).
Ophthalmologic follow-up will be determined by the referring ocular oncologist, depending on the treatments undertaken and the presence of a genetic predisposition. A genetic study for relatives free of the disease but potentially carrying the mutation is usually proposed, with the goal of establishing a specific ophthalmologic surveillance program.